
CJ Barnum spoke, I listened
- Carl Kestens
- Mar 18, 2022
- 23 min read

Updated: Mar 19, 2022

Introduction
CJ Barnum, head of neuroscience at INMB, has a talent for translating his superior scientific knowledge to a crowd. The below is a partial excerpt of a presentation he had presented on 17 August 2021, and which has now been shared on INMB's Twitter feed. I still find it the best introduction to INMB's XPro platform by far. Most striking to me were his first slide, the approach of inflammation as a low-grade disease, the link between TNF and amyloid and tau pathology, the dynamic character of the immune system, and an advice for cautiousness in interpreting any (non-downstream) biomarkers.
The science is extremely solid here, and that's what I love about INmune Bio, and the man has a lot to say. Reading should be shorter than listening in this time-consuming world, hence this article. Of note: this was prior to the strong September readout, again confirming where they will be taking this drug. I have left out some parts including the forward-looking statements. Without further ado....

Introduction of CJ Barnum by Prof. Malù Tansey: [...]
We started looking at the dependence on soluble TNF as a cytokine, and he was one of the first people who actually looked at how this one biologic we had been looking at with central delivery might actually cross into the brain when given peripherally. And so that changed the landscape for us, and we started looking at how maybe the role of inflammation peripherally could affect the brain, and so from then on we started looking at how peripheral triggers of inflammation could really drive central disease, and he authored several key reviews for us 10 to 12 years ago really, before inflammation was hot. One of them was called ‘peripheral inflammation: the dark passenger in Parkinson's’, and so I think he was ahead of his time. […]
CJ Barnum: […]
The one thing I want to talk about is: you know, immune dysfunction is probably the most important disease, and it doesn't even exist. We know that it accounts for at least 50 percent of all mortality, and there's some estimates that account for 80 percent of all morbidity. Yet we don't recognize it as a disease. I think what's one of the interesting things scientifically speaking that has come out of Covid is that we now recognize that the immune system can have a dramatic effect on how we survive and respond to different stimuli, and I think that's actually madea difference for us in that regard.

So what causes immune dysfunction?
Well virtually anything the immune system is in charge of taking care of the entire organism. It responds to any perturbation, any deviation from homeostasis.

This is an example from Furman et al. in 2019, and basically what they have here is just a few things here that ultimately cause disease and the consequences of that, and in the middle they have this gear here and these are pro-inflammatory cytokines. These are things like TNF and IL-1, these are largely driven by the innate immune system.
When the innate immune system becomes dysfunctional you get downstream changes and the adaptive immune system, and it really is tied to every single disease we see. I think the one thing we don't have in here is genetics, and the interplay between these sorts of things.
So, what does immune dysfunction look like?

Over here on the left what you see is a normal inflammatory response. So, asthe stimulus occurs, the immune system responds, then over time it resolves in a meaningful way. With chronic inflammation you can have a couple different scenarios. You can get a hyper response, you can get a hypo response, or you can get a response that just doesn't resolve in time. And this really what we define as is immune dysfunction or chronic inflammation.
Typically this response looks a lot like what we see in inflammatory aging, where you'll have a response and it just doesn't resolve all the way. So you've got this low grade response that does not resolve over time and as time goes on it gets worse and worse.

So, I just want to make one comment here. Probably the most important lesson I learned in graduate school about the immune system, especially the immune system in the brain, is it does not care about preservation of function. It only cares about survival, and it will take a nuclear warhead to a fistfight every single time. And I think a good example of this, besides coronavirus, is what you see in things like ischemic stroke or TBI, where you have this core injury and this penumbral region and this penumbral region is obviously driven by pro-inflammatory inflammatory cytokines. The idea is just to make sure you kill everything, but it's because of this we lose function over time, and we don't get the same function that we need. And I think this is a key element.
So how does this relate to Alzheimer's disease?

I'm going to talk about how we have looked at this in Alzheimer's disease. I'm going to give you a little bit of history about this, and then tell you how we're pursuing it. So we know that Alzheimer's disease actually suggests that it's an immunologic disease.

If you look at the genes associated with Alzheimer's disease, the high-risk genes: more than 60 of them are in what we believe is the innate immune system. Many in microglia, astroglia and oligodendrocytes. But when we look at this from a what's happening, what it's telling us is that it's involved in every single aspect of innate immune function. So you get things like cytokine secretion phagocytosis impairment and trophic support lysosomes and migration.
And the point here is that there are multiple ways to get to a dysfunctional immune system. So from a drug development perspective, well we would like to look at the genetics and say for example: TREM 2, we need to target TREM 2. But the genetics is really telling us that the immune system is dysfunctional.

And what we should be thinking about is a little more broadly about: what do the genetics mean? How do we put that into context, and try to find therapies and targets that are more likely to affect the immune cells more broadly? So, one of the things that drives me crazy is: every time I go to a talk and somebody says “the greatest risk for developing Alzheimer's disease is age”, that doesn't mean anything to me as a drug developer, because I don't know how to intervene that. But I'm here to argue that what I'm thinking of is that age is equal to inflammation, and here's an example of this.
We know by the time that you've reached your ‘30sand ‘40s, your immune system starts to become dysfunctional. We have entire fields surrounding this.

And what this graph is showing from a group at Duke in 2018-2019 is that age accounts for more than 80 percent of the variance and change over time. And so, I'd like to think that when we start talking about age what we're really talking about is dysfunctional immune system that has gone awry. So if that's the case, there should be at least some clinical evidence that the immune system is involved and targeting the immune system is neuroprotective and the answer is: there is.

So here's some data from a group at Brown that was published in 2016 looking at the risk of Alzheimer's disease in patients with chronic inflammatory disease. And what they found is that, if you had rheumatoid arthritis, your risk for developing Alzheimer's was 800 percent greater than the average person in the general population, unless you're on a TNF inhibitor. If you're on a TNF inhibitor, your risk was 60 less. The important thing here is this was only a TNF inhibitor so there are differences between classes of anti-inflammatory or immunosuppressive drugs, and again this is eight and a half million claims.

In 2019 a group out of Cincinnati took this a little further and looked at over 56 million claims, and what they found was that regardless of the disease, the chronic inflammatory disease, if you're on a TNF inhibitor, your risk for Alzheimer's was 50 to 70 percent less than the average population.
This is a pretty big difference.

Of course if you look at human studies you can see that there's plenty of evidence that TNF and other cytokines as well are increased in both the plasma, the CSF, we know it co-localizes with plaques in the brain and it correlates with disease progression.

For those of you that are interested in amyloid and tau, what's very interesting about this is, there is a TNF promoter on the APP gene, and the gene that drives beta-secretase. It facilitates alternative processing of beta-secretase and increases the likelihood of the development of amyloid plaques.

We see the same thing with tau. TNF increases neuro-accumulation within the neuron. It drives tau hyperphosphorylation and promotes neuronal cell death, and so all these data really support the idea of looking at TNF. But we've actually shown in the animal models looking at our drug that regardless of the animal model that we've looked at, we see a very broad improvement, and by blocking TNF and synaptic dysfunction we show cognitive improvement.

And in this case you're seeing a spatial learning test. I think one of the the most important things I like to discuss is that if you look at immune dysfunction, if you normalize the innate immune system, in this case we're using our novel TNF inhibitor that I'll tell you about a little bit in just a moment, you can normalize the adaptive immune system.
I think this is a really important point.
The adaptive changes, webelieve, are downstream of innate immune dysfunction. And of course, if you wanted any evidence that inflammation is important for amyloid pathology, we've got data in three different transgenics, amyloid overexpressors that show, if you give a TNF inhibitor, you can prevent the development of amyloid pathology.
So, that's great and that's wonderful, but why do we care? There are plenty of anti-inflammatories on the market and why are they not effective ?

The answer is: because how you target the immune system is ás important if not more important than just targeting the immune system.
What we have here on the right is a little schematic of immune cells that are supporting the neurons. I actually like to think of neurons as infants. You know, infants can't feed themselves they can't bathe themselves, they can't socialize themselves ,they can't do anything on their own, they require parents. That's what the immune cells in the brain are. They provide all the stuff necessary to communicate, so by suppressing those, you're putting those neurons at a disadvantage.
Of course you want to target what we call the chronic inflammation, the destructive aspects of the immune system, when they become dysfunctional.
And so what I'm going to tell you about now is our therapy which we believe is a first in class, that is selectively targeting the bad, and by default promoting the good.
So how does this work?
So the drug itself is a next generation TNF inhibitor and to understand this you have to understand a little bit about the biology.
TNF comes into the world as a transmembrane protein.

And as a transmembrane protein there are three monomers that trimerize to make this protein. Those trimerized proteins can initiate signal transduction via the TNF R1 or R2 receptor. During times of stress or chronic inflammation, this becomes cleaved, and you get these soluble monomers. It's these soluble monomers again have to trimerize, and then they can bind to receptor. It's the soluble TNF that's driving the destructive chronic inflammatory aspects of inflammation, and the transmembrane that's actually driving the good, and I'll talk about that in a moment.

So the current drugs on the market block both. So you get the benefits of reducing inflammation by blocking the soluble TNF, but you also get the liabilities associated with that. So how does this drug work?
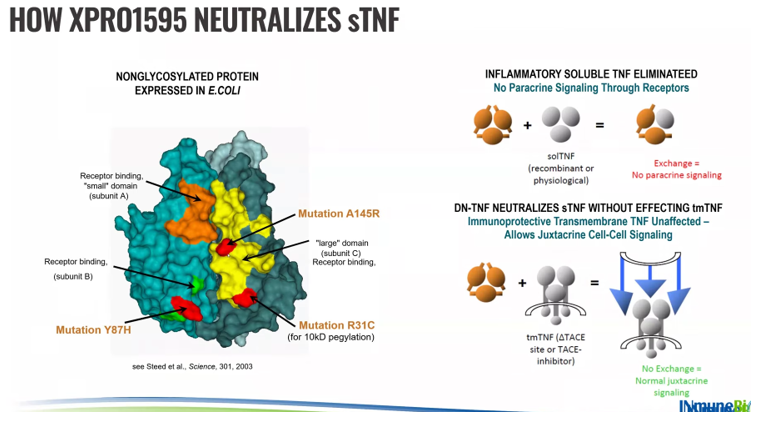
This is an extremely elegant drug.
It is identical to the TNF that you and I make, with the exception of a few point mutations. Some of these mutations are in the receptor binding domain, and one is for a pegylation to extend the half-life of the drug. And you can see normal TNF signaling requires a homotrimer. This drug can freely exchange with the native TNF and form these heterotrimers in a two-to-one or one-to-two configuration, and because the mutations are in the receptor binding domain, they cannot bind to receptors, so they neutralize the soluble TNF.
Right, and as a result it stops the progression, the feed forward aspect of inflammation, and promotes towards the transmembrane, because the transmembrane protein down here in the far right is anchored to the stock. It cannot interact, and so transmembrane remains intact, and the significance of this can be seen in an animal model of MS.

So here's an example of normal myelin staining. This is the corpus closum. If you give animals cuprizone which is one way to induce demyelination in animal models, you can see a loss of myelin; these little shark bites here on each side. If you give a non-selective TNF inhibitor you can see that demyelination actually gets worse, behavior gets worse.
This is not just an experiment. Back in the 90’s, Roche developed a compound called Lenercept. Lenercept, like all the other TNF inhibitors, is non-selective, so it blocks both transmembrane and soluble TNF, and they had to stop the study, and the reason they had to stop the studies was because patients got worse. This is exactly what happened. In fact, right now, if you look at some of the FDA reporting evidence, the most common cause of late life MS is chronic use of TNF inhibitors.
But it turns out, if you selectively neutralize soluble TNF, not only do you not get demyelination but you actually promote myelination. So the difference is extremely important as it relates to CNS disease, and to give you just a scorecard here of what this means.

Both the non-selective and this drug XPro1595 will decrease inflammation but XPro is not immunosuppressive. It does not demyelinate. It is neuroprotective and it enhances neuroplasticity.

So with this data we applied to the Alzheimer's association for a cloud grant. We were fortunate enough to receive that grant two years ago, and the goal of the study really was to demonstrate that we could give this biologic peripherally and reduce biomarkers of neural inflammation. And this study was a little bit different. It was a 12-week study but we selected patients based on the diagnosis of AD, and whether or not they had biomarkers of inflammation. So there's two key things here. I did not care what stage of disease they were in. All I cared about was they have biomarkers of inflammation and these are the ones that we chose.
And I'll talk about that in just a moment. But the goal here was: after 12 weeks, could we see a change in the biomarkers of neuroinflammation? And we looked specifically at neuroimaging and cerebral spinal fluid. So let me talk a little bit about bio. This is one of my digressions of biomarkers of inflammation because I get asked all the times about those, because inflammation is obviously quite prevalent.

When I think of biomarkers of inflammation, I think of two different versions of it. I think of behavioral biomarkers of inflammation and I think of the normal typical biomarkers of inflammation that we're used to measuring. But let's say a patient comes in and they present with a disease, but the disease doesn't fit into a nice neat bucket. Right, you've got all these checkboxes, but they've got all these non-specific symptoms, and they present with things like depression, anxiety. This is the concept of sickness syndrome. So sickness syndromes are adaptive behaviors designed to conserve and redistribute resources during times of infection. These sickness syndromes are facilitated by pro-inflammatory cytokines like TNF and IL-1, and of course resolve once the infection is dispatched. However during chronic inflammation and immune dysfunction, those inflammatory cytokines remain high enough for a long enough period of time that these sickness behaviors turn into a depressive episode. And so many times patients will present with things like depression, anxiety, neurocognitive symptoms, melancholy , psychosomatic and such. And these are are really good clues that the patient has some underlying inflammation as part of their biology. And so the next thing I would say is: inflammation has been, I think misunderstood quite a bit, and I think the reason for that is we look at it especially as neuroscientists.

We look at it through the lens of the neuron, of how we were taught to look at neuroscience, where we take a disease and we take a control, and we see a difference. Right, we know, in an Alzheimer's patient they'll have amyloid plaques, but a control patient will not. Of course, that's not entirely true, but in theory that's the case. That's not how the immune system works. The immune system is not static. It’s much more dynamic, and so if we look at 10 studies, we may see that controls versus disease, in this case this is ALS, are different 50 of the time. But the right comparison is actually: inflammation or no inflammation. Because many of these patients, because inflammation is so pervasive have other diseases where inflammation is driving it. I think the thing to remember is the immune system, and chronic inflammation, immune dysfunction goes after your weakest biological link whatever that may be.

And that's going to be different for everyone. So the right comparison is: do they have inflammation or not, and that's just not the way that we're thinking about it. The other thing to think about is that the immune system is really really dynamic. so here's an example of a healthy animal treated with a polysaccharide.

So this is a component of gram-negative bacteria, it's non-infectious, and what you can see is over the course of, I think this is a four-hour period, that you start to get an increase in cytokines and then the immune cells start to come in at different times. So, if we look at it from trying to determine where what you know what mechanistically is driving it, our conclusion would be determined by where we're measuring this in relation to time. So if we pick it right here, we'd say, well it's the microglia, and if we looked right here, we would say it's the neutrophils, and you have to think about if you've got thousands of patients or hundreds of patients in a clinical trial that you're treating the time frame of this is going to be very different. So what we're looking can be particularly confusing, because there's no real solid stable measure, if you will.
Even if we look at cytokines themselves, and this is another thing, we see all the time again healthy animals treated with LPS. If we look over the course of four hours, first thing that goes up is TNF. It's the master cytokine, it drives these downstream changes. Then we see changes in IL-1, IL-6 goes up and stays a little bit longer before we start getting some of these inflammatory cytokines.

So again, where you look after a stimulus is going to tell you what is what is elevated. From our perspective, weare interested in patients that have inflammation, that don't have infection. So the specific inflammatory factor is not as important to me as evidence that you have inflammation. To make this even more complicated: measuring cytokines and chemokines is particularly difficult. There's an enormous circadian variation that can go up almost a hundredfold.

It's expressed at really low levels, so even pathological levels are expressed in the, let's call it, 20 to 40 picogram per ml. We know that blood and CSF don't correlate, so trying to understand how blood inflammatory cytokines relate to CSF inflammatory cytokines doesn't always work and of course it requires specialized instruments.
I think the thing that I've learned most from doing this for 15 or so years is that CRP, something that we're all familiar with that's used in the lab, is probably the best single inflammatory factor if you're looking at blood. It’s not subject to the same circadian variation, there is some but not to the same extent you get with cytokines.

It actually correlates almost perfectly with what you see in the brain, and obviously it can be measured in any lab worldwide. And so, if I go back to the biomarkers that we chose, we chose biomarkers that we know are more stable, easily measured in any lab in the world except for APOE4, and there's some evidence that they predict response to anti-inflammatory treatment.

And so in our study if they had one of those four biomarkers they were eligible to participate in our study. So, the other thing that I will say is, because there's so much instability and dynamics to it, our approach to measuring these biomarkers was a little bit different.

You know, we've got a TNF inhibitor, we know TNF inhibitors are potently anti-inflammatory. It should be obvious in multiple different assays and platforms, and so what we did is we looked at CSF, we looked at two different platforms, one a cytokine panel, and one of proteomics approach, and we also looked at MRI free water white matter, and let me tell you a little bit about that now.

So, white matter free water is a biomarker of neural inflammation and basically what it's doing is looking at the free water surrounding the white matter. You can think of it as edema or swelling. There's plenty of evidence that it's increased in Alzheimer's patients.

So you can see here from normal controls to MC to alzheimer's patients the neural inflammation is increasing by this metric. Uou can see here that over two years you're looking at AD patients,, the free water increases over time, you see a more stable free water neuroinflammation in MCI patients and in normal controls as well. I think what's also interesting about this is: free water actually correlates really well with amyloid over two years.
And so we've got this ability now to detect neuroinflammation not invasively using an MRI standard sequence that I believe adds about five minutes to a normal MRI scan. So what can we do with this. So we're working with a company called Imeka, and Imeka has pioneered this whole this tractography to free water and commercialized it, where we can look at whole brain free water, and they do this in a way, they call it a safe mask, where they can quantify the free water that's not contaminated by the ventricles. Soyou can now see where in the brain this is happening.

They can also look at specific white matter tracks, so they can identify ones that are important and relevant for the disease state that you're looking at, and this is what we have done with them.
So here's a data set that we released I guess it was a little more than a year ago now, where we only had six patients and we used an ADNI cohort for contacts, because we didn't have a placebo-controlled trial.

This is an open label study but generally speaking over the course of 12 weeks the ADNI cohorts increase about 5 percent, and you can start to see that we have a dose response in whole brain inflammation. When we start looking more specifically at white matter tracks, what we found was that irrespective of dose, we got a massive reduction in neuroinflammation within the arcuate fasciculus, so this is an area that connects Wernicke's, and Broca, has some pre-motor connections as well, and this is important for language processing, the ability to understand or express speech which is obviously relevant for Alzheimer's disease. Of course to validate these things you have to anchor them to something that we all know about.

So what I'm showing you here is cytokine data from this this Olink target 48, and you know, one of the things that, and what you're seeing is everything that is within the limit of detection. I think one of the things that always drives me nuts is when people report, you know, on we reduced inflammation, they show two or three cytokines or maybe just one cytokine and the question I always, you know,, how is that relevant to the mechanism of your drug, and did you look at anything else, andthe answer is they almost always looked at everything else, that was just the only one that was significant. So we believe that looking at the pattern of results is more informative than looking at any single inflammatory factor mostly because of what I showed earlier regarding the dynamics of it, and so you can see that across the map broadly we get a reduction in inflammatory factors. If we average this out we take a deposit, it's about a 15 percent reduction, and this is in patients with our high dose, we have multiple doses we tested but I'm going to show you mostly patients with a high dose because. That was the one that was most effective and we added a few more patients to this white matter free water, so we have six patients, and of course if you want to be able to argue that your metric is really reflecting no inflammation you need to show that it correlates with the type tried and true.

So, if we if we look at the correlation between this MRI free water and CSF inflammation via this composite, and these are patients irrespective of dose, you can see that even in nine patients we have a very nice correlation between that. So we think this can be used in the future to help us identify patients that either have biomarkers of inflammation, then we're getting pretty close to identifying a threshold at which we can now look and say: if they have a value of free water above x, this means they're probably inflamed, so that's great we can reduce neuroinflammation and we should have a TNF inhibitor. The question is, so what? Who cares, what are the consequences of that? So, just like, you know, targeting amyloid or targeting neural inflammation, you know, they're causing something downstream, it's leading to a downstream change in the brain. Right, what is happening with the brain, what is happening with the neurons?

And so, we've attempted to look at this again using multiple platforms.

In this case we looked at proteomics, and we looked at different metrics of imaging. Now, the one thing I will say about this is, when you start talking about looking at downstream changes, you really need to understand the biology of your drug, and the mechanism, and for us luckily, we know a lot about TNF and preclinical and clinical models.

We know that it contributes to every single aspect of AD pathology, and more importantly as it relates to our drug, we know a lot about our drug, we have 68 publications, more than almost 60 percent of them are within CNS. I think the really unique thing about this drug is that this is a drug that was licensed from a company that originally developed it, and it was put on the shelf back in 2006, and what's interesting about that is at that time the company gave the drug to whoever wanted to test it so except for maybe four published studies the rest of them were done by research scientists that were interested in understanding from a mechanistic point of view. They had no financial incentive for it, and so we've got this massive data collection that all shows and points the same thing, so we know a lot about our drug, we know a lot about the biology both within a disease and across different diseases as well so again this is important when you start looking at downstream changes and the consequences of that. So this is an unbiased report that we received from proteome sciences.

This is a company that that analyzes the disease state tissue, so in this case Alzheimer's brain tissue, in parallel with our CSF samples, and this allows us, this gives us greater sensitivity and specificity to identify those proteins that are most relevant for Alzheimer's disease and those that are expressed at very low levels, and I think the most important take-home message for me is, when I receive this report, this reads exactly as I would expect based on our animal models.
So we know that the drug has important effects on the immune system microglial activation immune function. More broadly, we know it has an important impact on synaptic proteins and axonal proteins and dysfunction, and when they looked at the functional analysis, they could see this in really good detail.

So, we don't have the full report available, but let me just give you a snapshot of some of the proteins that are relevant in different classes.
So what we see is, we see a very large change in immune related proteins.
So we see a reduction in c-reactive protein. We actually see a really large increase in YKL-40, which is a which an immune protein that is tied mostly to astrocytes, although the significance of that I think remains to be determined. I've got two examples of synaptic proteins, so contactin-2 is important for neurite outgrowth and extension on the presynaptic neuron, and we get a big increase in that, 222 percent. Neurogranin: as neurogranin increases, it reduces synaptic outgrowth and extension from the postsynaptic cell, and we get a decrease there, so that's exactly what we would see, and if we look at two neuronal injury markers, we can see a decrease in both neurofilament and vilip-1 as well, so these proteins are doing the changes that we're seeing, are completely consistent with what we see in the animal models and we in such a very small number of patients.

The other thing that we can do with these proteomics is, we can start looking at dose response. So what you're seeing here is a volcano plot, and each dot represents a protein, that was significantly changed in one direction or another by one of the two doses. All the green dots reflect proteins that were significantly regulated by both of these two doses, and the red ones just by the high dose. What's interesting about this is, if you look at these proteins, these are the CNS related proteins, looking at that are relevant for Alzheimer's disease, so we can use these proteomics to help us identify a dose response with that.
So the last thing that I want to talk about is some of these new MRI metrics that we are we are using, that we think are extremely promising.

You know right now, the traditional way to look at this is looking at volumetric changes, but those are, you know, fairly insensitive to changes, even within a year, and in the drug development world, that's a very long time, but there's plenty of information that is embedded within the new imaging and DTI and that sort of thing that can be extrapolated, and so we're working with two companies. Again this company called Imeka looking at white matter quality, and a company called Oxford Brain Diagnostics looking at gray matter. I'm not going to show you the gray matter data today for simplicity's sake, but let me just tell you what it measures. So obviously in normal healthy brain tissue you have these nice organized columns with these local connections, and in a disease patient, you actually can see that these things are not as organized and you're losing local connections.
It turns out that this doesn't correlate with volumetric changes, but it does correlate really well with clinical decline, and so this metric is maybe a little more useful as it relates to looking at changes over time, that are a little more sensitive than we get with volumetric assays, we have become more interested in looking at white matter so axe you know myelinated axons in this local environment, you can see normal healthy white matter you've got lots of axons they're myelinated they've got you know a reasonable amount of free water in the space, and supporting immune cells, and when the patient becomes disease, you start to lose axons, you start to lose myelin and all sorts of things and you can measure this we call it something called apparent fiber density. So what is apparent fiber density? So here's a micro pictograph of what you would expect in healthy white matter. The blue would be water free water, the green would be the axons, and the purple would be myelin, and you can see you've got lots of axons they're nice myelinated with a little bit of free water.

There's an animal model of Alzheimer's. You can see, what happens, is you're losing axons. These axons are actually swelling, which is what typically happens before they burst, and you lose them, and the myelin you can see is not compacted, it's starting to get lost, and so we can actually capture that subunit, this sort of this environment here, this cross section, using this apparent fiber density metric, and so I think one of the things that was really interesting and important to us was that we could see pretty big improvements in this white matter change within a very short period of time.

For contextual purposes again if you look at an ADNI cohort, you see a reduction in white matter that sort of plateaus around 12 months, probably because you've sort of reached the plateau of white matter loss in the brain, but by three months we get a 10 percent improvement that goes up to 16 percent by nine months, in just six patients, and this is this is actually within that same region, the arcuate fasciculus, which improved again 10 percent within these three months. I think the take home message here to add to that is, the company looks at about 35 white matter tracks that they've identified as most important for AD, and we get about we get an increase in apparent fiber density in 34 of those 35 white matter tracks and this is an average of six to twelve percent depending on the bundle and these correlate very strongly with CSF markers.

So I know I've given you a lot, let me just sort of summarize briefly. I think you know one of the things that is really important is, we need to start thinking about immune dysfunction as a disease and what it means across all the different CNS diseases, because it accounts for so much mortality and even more mobility I think.
The other thing to think about is that the dysfunctional immune system really does go after your weakest biological link. There are five of us sitting in a room, and we've all got immune dysfunction that's going to manifest a little bit differently, as well biomarkers to identify patients that have inflammation both behavioral and biological, they're really readily available and quite honestly, if you had to pick one, CRP is is pretty good but CRP is also cyclical over the course of 12 months. It will wax and wane over time as well, so you have to be aware of that. We don't have biomarkers that identify what I would call the trait of immune dysfunction, this is somebody who has a dysfunctional immune system but currently is not displaying signs of inflammation. We are working on that, we need those sorts of things, I think that will be really useful moving down the road and again to reinforce these these inflammatory biomarkers of treatment response. They're really more complicated because the dynamic nature, and you really want to move downstream into something that's a little more reliable but that also requires you to understand your biology very well.



The reason that we have not found a clinical solution to Alzheimer’s is that we didn’t understand the disease. This man understands the disease better than anyone I’ve ever been exposed to. He also has the drug to modulate the disease process. Like every other disease that we have conquered ( polio, small pox, etc), it was done by methodical, relentless, and clear eyed research and perseverance. Which obviously he is doing. Not by throwing things against the wall and seeing what sticks (which is what Alzheimer’s research has been up until now)